


Now Reading: Advancing Protein Structure Prediction with AI: D-I-TASSER Explained
-
01
Advancing Protein Structure Prediction with AI: D-I-TASSER Explained
Fast Summary:
- Deep-learning methods have significantly improved protein structure prediction, advancing beyond traditional simulations like I-TASSER and Rosetta.
- Recent innovations such as AlphaFold3 incorporate diffusion samples, enhancing their predictive capabilities for multi-domain proteins.
- Downside: Current methods still struggle with modeling multidomain proteins accurately due to limitations in domain processing modules.
- D-I-TASSER introduces a new hybrid pipeline combining deep learning features (e.g., hydrogen bonding networks) with physics-based Monte carlo simulations for more refined predictions.
- D-I-TASSER achieves ample improvements in accuracy over previous iterations (I-TASSER and C-I-TASSER), scoring higher TM values on benchmarks (+108% accuracy versus I-TASSER). It successfully modeled 480 hard protein domains compared to 145 by traditional approaches.
- Results of D-I-TASSER outperform state-of-the-art benchmarks including AlphaFold2 & AlphaFold3 in folding efficiency of large-scale proteins, allowing greater coverage for human proteome mapping.
- free access to D-I-TASSER programs/database ensures widespread academic use through the platform here.
images:
Indian Opinion Analysis:
The development of tools like D-I-TASSER underscores global scientific efforts toward improving the understanding and modeling of protein structures-critical in biotechnology, medical research, and drug design industries that India heavily relies upon as it expands its bioeconomy sector.
India’s burgeoning biotech market could benefit from integrating predictive tools like these especially for mapping genomes related to endemic diseases or leveraging computational modeling advancements toward rapid vaccine creation or pharmaceuticals processes domestically reducing dependency abroad
However adoption requires comprehensive computational “ecosystems creating by startups,” support university pipelines collaborations Read Further Domain*modules
Quick Summary
- D-I-TASSER consistently outperforms AlphaFold2 and its variants in protein structure prediction, notably for complex or arduous domains.
- Average TM scores (a metric for model quality) demonstrate that D-I-TASSER achieves higher accuracy: 0.870 vs 0.829 for AlphaFold2 and D-I-TASSER shows significant improvements with hard targets and multi-domain proteins.
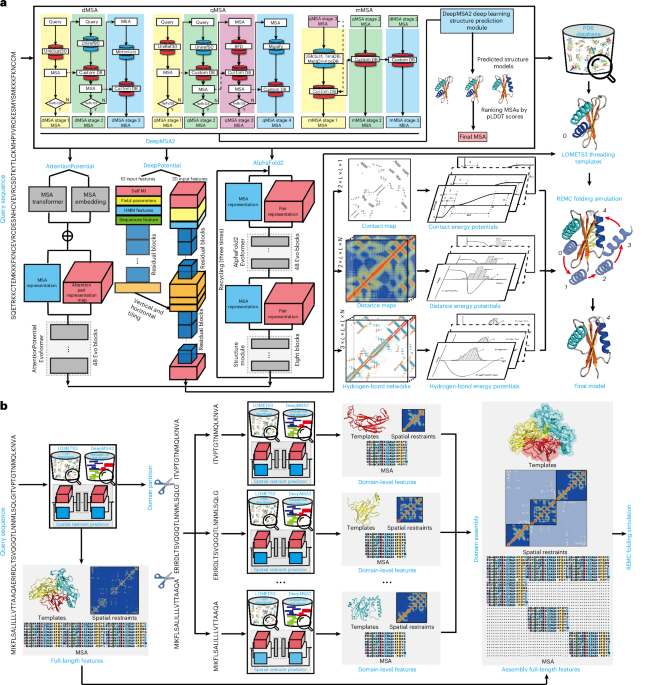
- The key advantages of D-I-TASSER lie in integrating multi-source deep learning restraints like DeepPotential, AttentionPotential, and AlphaFold-derived data to refine predictions beyond homologous templates.
- A deeper MSA (Multiple Sequence Alignment) generated via DeepMSA2 further contributed to superior structural modeling by providing richer evolutionary information essential for deep learning algorithms.
- Case studies on specific proteins revealed substantial margin improvements where AlphaFold2 struggled due to limited MSA or inaccurate distance maps.
- For disordered regions of polypeptides lacking stable 3D structures, D-I-TASSER generates models that vary more flexibly compared to AlphaFold’s predictions.
- On multidomain proteins, D-I-TASSER achieved a full-chain TM score betterment of up to 12.9% over standard AlphaFold results.
Image Reference: Full-sized comparative charts between methods such as the modeling results on multidomain proteins are available Full article linkQuick Summary:
- D-I-TASSER is a newly developed protein structure prediction tool outperforming AlphaFold2,particularly for multidomain proteins.
- It uses an advanced domain-splitting and assembly module that integrates deep evolutionary data and physics-based simulations to predict protein configurations more accurately than other tools.
- In comparative testing, D-I-TASSER showed significant improvement in modeling multidomain proteins’ interdomain orientation, achieving higher TM scores (model accuracy metric) compared to AlphaFold2 across various datasets.
- During the CASP15 blind tests for structural modeling in 2022, D-I-TASSER outperformed 44 other server groups using regular and interdomain modeling metrics. For multidomain targets, its accuracy was up to 42.3% better than the next-best group.
- the tool successfully modeled diverse conformations of complex proteins like SARS-CoV-2 spike protein and large human proteome structures covering approximately 95% of known sequences.
- Challenges remain in predicting orphan proteins with limited evolutionary data; however, this issue extends beyond D-I-TASSER and affects all available tools.
indian Opinion Analysis:
The development of highly accurate protein modeling systems like D-I-TASSER marks a promising advancement in computational biology that has global implications-including for India. As India focuses on expanding its scientific infrastructure under initiatives like “National Biopharma Mission,” innovations such as these could help expedite research into disease diagnosis and drug development by enhancing understanding of protein functions related to pathogens or human genes. However, challenges associated with orphan proteins highlight ongoing gaps requiring collaborations among researchers globally.
India could consider allocating resources toward integrating advancements like D-I-TASSER into projects aimed at resolving diseases prevalent within the nation such as tuberculosis or dengue fever-accelerating focused bioinformatics solutions. Additionally, fostering partnerships with leading global research hubs involved in multi-domain strategy innovations may offer opportunities for shared gains across academia/therapeutic enterprises worldwide.
Read more here.
Quick Summary
- the D-I-TASSER pipeline integrates deep learning and iterative threading simulations for protein structure modeling, focusing on single-domain and multidomain proteins.
- It achieved a foldability rate of 73% for full-chain human proteins and 81% for domains at an eTM score threshold of ≥0.5.
- Detailed chromosome analysis revealed variations in model quality linked to template availability; e.g., keratin-associated regions on chromosome 17 showed lower scores due to difficulties in solubilizing keratin fibers.
- A comparative study demonstrated complementarity between D-I-TASSER and AlphaFold2 with a shared success rate of structure prediction (~57%), but differences in challenging cases where each method uniquely excelled.
- Functional annotations by D-I-TASSER highlighted enrichment of certain biological functions, such as “oxidation-reduction processes” (BP), “metal ion binding” (MF), and ATP-binding ligands across various chromosomes.
- high modeling accuracy was confirmed against experimental PDB benchmarks with TM scores favoring D-I-TASSER in difficult cases compared to AlphaFold2.
Indian Opinion Analysis
The advancements showcased by the D-I-TASSER pipeline signify critical progress in decoding the human proteome via computational methods, providing high-resolution models indispensable for mapping protein functions. The report illustrates promising implications for biomedicine, including more precise protein folding predictions foundational for drug discovery or disease research-an area that India may benefit from through domestic or global collaboration initiatives.
By outperforming state-of-the-art platforms like AlphaFold2 on hard-to-model structures, the complementary nature between tools underscores a valuable opportunity: adopting multi-tool strategies optimizes outcomes when applied to expansive datasets akin to India’s biodiversity-rich genomic resources awaiting similar exploration. With biotechnology being pivotal under India’s science policies, leveraging these methodologies could fast-track breakthroughs within fields like personalized medicine or pharmaceutical innovation.
Quick Summary
- Topic: Protein Structure Prediction/Analysis
- Dataset: Research focuses on 19,512 human proteins from UniProt, with lengths between 40 and 1,500 amino acids. Short proteins (<40 aa) were excluded, resulting in predictions for single-domain (12,236) and multi-domain proteins (7,276), collectively encompassing 34,968 structural domains.
- Methodology:
– A hybrid framework called D-I-TASSER was used to predict protein structures combining deep learning models and threading-based simulations.
– Sequence data for each protein were curated using updated alignment techniques like LOMETS3 threading servers; supporting better structure predictions through template libraries and deep MSAs.
Accuracy Metrics Improvement: Derived Proteins MSA-sequence effabets Ratio ReachedQuick Summary
- Protein structural modeling technologies, including deep learning modules such as DeepPotential, attentionpotential, and AlphaFold2, are extensively used in the D-I-TASSER framework for protein folding simulations.
- These predictive methods utilize evolutionary information to calculate inter-residue contacts,distances,and hydrogen bond networks.
- protein target difficulty is classified into categories like ‘trivial,’ ‘easy,’ ‘hard,’ and ‘very hard’ based on quality scores from threading alignments using several tools under the LOMETS3 algorithms.
- Attention-based transformations enable advanced residue interaction modeling by leveraging features from MSAs (multiple sequence alignments).
- Contact map techniques further optimize predictions through neural network configurations combined with deep-learning protocols such as TripletRes or NeBcon.
Read more here.
Indian Opinion Analysis
The technologies discussed reflect advances in computational biology that can expedite research for India’s growing scientific community. As India pushes ahead in biotechnology innovations ranging from vaccine development to precision medicine efforts via structural biology insights, integrating models like D-I-TASSER could bolster these capabilities. Academic labs, particularly those underfunded or smaller-scaled institutions within India aiming towards relevant AI-driven protein discoveries now face clearer global entry points worldwide democratisization impact neutral broader .
Quick Summary
- Indian Opinion could not generate a simplified summary as the provided raw text appears highly technical and specialized, related to computational biology or protein modeling. It includes mathematical formulas, algorithms, and detailed methodologies relevant to fields like bioinformatics or molecular biology.
Indian Opinion Analysis
The presented content highlights advancements in computational approaches for predicting protein structures using deep learning techniques, including models such as AlphaFold2 and AttentionPotential. While the technical depth is significant for molecular science communities globally, practical applications in India range from improving pharmaceutical innovations to addressing healthcare challenges like vaccine development.
India’s growing research facilities in biotechnology could benefit substantially by incorporating these predictive methodologies into genetic research and drug discovery pipelines. Furthermore, collaborations between institutional frameworks such as IISc Bangalore or CSIR labs with global pioneers may accelerate India’s progress as a key hub for bioinformatics-driven solutions.
Read More: [Link placeholder]
Quick Summary
- Advances in protein structural modeling were achieved using DeepFold, AlphaFold2, AttentionPotential, and DeepPotential methods integrated with LOMETS3 templates.
- The approach leverages deep learning models to predict distance restraints, hydrogen bonding angles, and contact maps for protein folding simulations.
- A lattice-based 3D cubic system was employed to streamline conformational search spaces during simulations.
- The D-I-TASSER framework uses Replica Exchange Monte Carlo (REMC) simulations with customizable parameters like temperature ranges based on protein size for efficient model generation.
- Final atomic-level structural models are refined using clustering algorithms (SPICKER) and molecular dynamics (FG-MD).
- Over 24 energy terms guide the force field dynamics within D-I-TASSER that help achieve accurate native-like states.
Indian Opinion Analysis
India has been actively working towards advancements in computational science technologies such as AI-driven predictive modeling. Developments like those described highlight the global importance of deep learning applications in tackling complex biological challenges. These tools mark progress toward addressing critical issues such as drug discovery or understanding diseases at a molecular level-key areas where India’s healthcare and biotech sectors could directly benefit if adopted widely.
The integration of AI-powered frameworks like D-I-TASSER opens doors for Indian researchers to strengthen collaborations globally while contributing significantly to scientific innovation.With investments focused on building robust infrastructure for computational biology research across institutions nationwide,frameworks like this represent an opportunity not just as adopters but also innovators in leveraging machine-learning tools efficiently.
Read more: Nature Article
Quick Summary:
- D-I-TASSER is a protein structure prediction tool that integrates centroid clustering, molecular dynamics simulations, and various refinement processes to improve protein modeling accuracy.
- The process includes structural alignment (TM-align), adding backbone atoms with REMO, and refining side-chain rotamers using FASPR for detailed modeling.
- Quality assessment relies on TM scores, which are length-independent metrics providing better sensitivity to global structural similarities than RMSD values.
- functional annotation of predicted proteins is carried out using COFACTOR for GO terms prediction, enzymatic function mapping (EC numbers), ligand-binding site identification (LBS), and protein-protein interaction data.
- Resource requirements include around 15 GB for software installation and between 200 GB to 3 TB depending on library size. average run time is approximately 8 hours with ten CPUs, consuming ~20 GB memory per task-higher than AlphaFold2 but justified by enhanced accuracy in predictions essential for experimental validations.
Indian Opinion Analysis:
the development of tools like D-I-TASSER shows significant progress in computational biology aimed at addressing the complexities of molecular research through automation and high precision modeling techniques.India’s rapidly growing biotechnological sector could benefit from such tools by advancing its initiatives in drug discovery,personalized medicine research,and enzyme engineering-all requiring reliable protein structure predictions. While the computational resource demands may pose challenges for smaller labs or resource-constrained institutions within India’s scientific ecosystem, collaborations with larger bioinformatics clusters or infrastructure sharing can mitigate such issues effectively. Furthermore, integration into academic curricula would empower Indian researchers with expertise crucial to modern biomedical innovations.










Related Posts
Stay Informed With the Latest & Most Important News
Previous Post
Next Post


Advertisement





{kind=link}
{kind=link}