Now Reading: Prime editor-based high-throughput screening reveals functional synonymous mutations in human cells
-
01
Prime editor-based high-throughput screening reveals functional synonymous mutations in human cells
Prime editor-based high-throughput screening reveals functional synonymous mutations in human cells
Data availability
The plasmids used in this study were deposited to Addgene or are available upon request. All raw sequencing data were deposited to the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation and Beijing Institute of Genomics, Chinese Academy of Sciences under accession number HRA007615. The human reference genome used in this study is GRCh38.p14 from the NCBI (GCF_000001405.40). Databases involved in this study included ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), SynMICdb (https://synmicdb.dkfz.de/rsynmicdb/), DepMap (https://depmap.org/portal/), GWAS Catalog (https://www.ebi.ac.uk/gwas/) and the Medical Research Council Integrative Epidemiology Unit OpenGWAS (https://gwas.mrcieu.ac.uk/). Source data are provided with this paper.
Code availability
The ZFC-eBAR (version 0.2.0) algorithm and DS Finder (version 0.1.0), implemented in Python 3, can be downloaded from GitHub (https://github.com/UronicAcid/ZFC-eBAR and https://github.com/UronicAcid/DS-Finder). Other processed data and code can be found on Zenodo (https://doi.org/10.5281/zenodo.14639522)69.
References
-
Kimura, M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 267, 275–276 (1977).
-
Shen, X., Song, S., Li, C. & Zhang, J. Synonymous mutations in representative yeast genes are mostly strongly non-neutral. Nature 606, 725–731 (2022).
-
Kruglyak, L. et al. Insufficient evidence for non-neutrality of synonymous mutations. Nature 616, E8–E9 (2023).
-
Cuevas, J. M., Domingo-Calap, P. & Sanjuan, R. The fitness effects of synonymous mutations in DNA and RNA viruses. Mol. Biol. Evol. 29, 17–20 (2012).
-
Kristofich, J. et al. Synonymous mutations make dramatic contributions to fitness when growth is limited by a weak-link enzyme. PLoS Genet. 14, e1007615 (2018).
-
Walsh, I. M., Bowman, M. A., Soto Santarriaga, I. F., Rodriguez, A. & Clark, P. L. Synonymous codon substitutions perturb cotranslational protein folding in vivo and impair cell fitness. Proc. Natl Acad. Sci. USA 117, 3528–3534 (2020).
-
Lebeuf-Taylor, E., McCloskey, N., Bailey, S. F., Hinz, A. & Kassen, R. The distribution of fitness effects among synonymous mutations in a gene under directional selection. eLife 8, e45952 (2019).
-
Sauna, Z. E. & Kimchi-Sarfaty, C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 12, 683–691 (2011).
-
Supek, F., Minana, B., Valcarcel, J., Gabaldon, T. & Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 156, 1324–1335 (2014).
-
Buske, O. J., Manickaraj, A., Mital, S., Ray, P. N. & Brudno, M. Identification of deleterious synonymous variants in human genomes. Bioinformatics 29, 1843–1850 (2013).
-
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
-
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
-
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
-
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
-
Nelson, J. W. et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 40, 402–410 (2022).
-
Chen, P. J. et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184, 5635–5652 (2021).
-
Zhu, S. et al. Guide RNAs with embedded barcodes boost CRISPR-pooled screens. Genome Biol 20, 20 (2019).
-
Zhu, S. et al. Genome-wide CRISPR activation screen identifies candidate receptors for SARS-CoV-2 entry. Sci. China Life Sci. 65, 701–717 (2022).
-
Sharma, Y. et al. A pan-cancer analysis of synonymous mutations. Nat. Commun. 10, 2569 (2019).
-
Kolde, R., Laur, S., Adler, P. & Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 28, 573–580 (2012).
-
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023).
-
Mathy, C. J. P. et al. A complete allosteric map of a GTPase switch in its native cellular network. Cell Syst. 14, 237–246 (2023).
-
He, Y. et al. Near-atomic resolution visualization of human transcription promoter opening. Nature 533, 359–365 (2016).
-
Kim, H. K. et al. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 39, 198–206 (2021).
-
Mathis, N. et al. Predicting prime editing efficiency and product purity by deep learning. Nat. Biotechnol. 41, 1151–1159 (2023).
-
Prokhorenkova, L., Gusev, G., Vorobev, A., Dorogush, A. V. & Gulin, A. CatBoost: unbiased boosting with categorical features. In Proc 32nd International Conference on Neural Information Processing Systems (eds Bengio, S. et al.) 6639–6649 (NIPS, 2018).
-
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548 (2019).
-
Wagner, N. et al. Aberrant splicing prediction across human tissues. Nat. Genet. 55, 861–870 (2023).
-
Barry, T., Wang, X., Morris, J. A., Roeder, K. & Katsevich, E. SCEPTRE improves calibration and sensitivity in single-cell CRISPR screen analysis. Genome Biol 22, 344 (2021).
-
Nackley, A. G. et al. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 314, 1930–1933 (2006).
-
Tuller, T. et al. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141, 344–354 (2010).
-
Bae, H. & Coller, J. Codon optimality-mediated mRNA degradation: linking translational elongation to mRNA stability. Mol. Cell 82, 1467–1476 (2022).
-
Dhindsa, R. S. et al. A minimal role for synonymous variation in human disease. Am. J. Hum. Genet. 109, 2105–2109 (2022).
-
Cuella-Martin, R. et al. Functional interrogation of DNA damage response variants with base editing screens. Cell 184, 1081–1097 (2021).
-
Hanna, R. E. et al. Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080 (2021).
-
Dujon, B. Basic principles of yeast genomics, a personal recollection. FEMS Yeast Res. 15, fov047 (2015).
-
Chamary, J. V., Parmley, J. L. & Hurst, L. D. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 7, 98–108 (2006).
-
Liu, Y. et al. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat. Biotechnol. 36, 1203–1210 (2018).
-
Radrizzani, S., Kudla, G., Izsvak, Z. & Hurst, L. D. Selection on synonymous sites: the unwanted transcript hypothesis. Nat. Rev. Genet. 25, 431–448 (2024).
-
Kudla, G., Murray, A. W., Tollervey, D. & Plotkin, J. B. Coding-sequence determinants of gene expression in Escherichia coli. Science 324, 255–258 (2009).
-
Gu, W., Zhou, T. & Wilke, C. O. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput. Biol. 6, e1000664 (2010).
-
Yan, J. et al. Improving prime editing with an endogenous small RNA-binding protein. Nature 628, 639–647 (2024).
-
Ren, X. et al. High-throughput PRIME-editing screens identify functional DNA variants in the human genome. Mol. Cell 83, 4633–4645 (2023).
-
Gould, S. I. et al. High-throughput evaluation of genetic variants with prime editing sensor libraries. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02172-9 (2024).
-
Cirincione, A. et al. A benchmarked, high-efficiency prime editing platform for multiplexed dropout screening. Nat. Methods 22, 92–101 (2025).
-
Belli, O., Karava, K., Farouni, R. & Platt, R. J. Multimodal scanning of genetic variants with base and prime editing. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02439-1 (2024).
-
Kim, Y., Oh, H. C., Lee, S. & Kim, H. H. Saturation profiling of drug-resistant genetic variants using prime editing. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02465-z (2024).
-
Herger, M. et al. High-throughput screening of human genetic variants by pooled prime editing. Cell Genom. 5, 100814 (2025).
-
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
-
Morales, J. et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 604, 310–315 (2022).
-
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
-
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G. & Neufeld, J. D. PANDAseq: paired-end assembler for Illumina sequences. BMC Bioinformatics 13, 31 (2012).
-
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
-
Xu, P. et al. Genome-wide interrogation of gene functions through base editor screens empowered by barcoded sgRNAs. Nat. Biotechnol. 39, 1403–1413 (2021).
-
Cleveland, W. S. Robust locally weighted regression and smoothing scatterplots. J. Am. Stat. Assoc. 74, 829–836 (1979).
-
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
-
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 (2004).
-
Hinrichs, A. S. et al. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 34, D590–D598 (2006).
-
Ding, B. et al. Noncoding loci without epigenomic signals can be essential for maintaining global chromatin organization and cell viability. Sci. Adv. 7, eabi6020 (2021).
-
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
-
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
-
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
-
Replogle, J. M. et al. Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol. 38, 954–961 (2020).
-
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
-
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
-
Robinson, M. D., McCarthy, D. J. & Smyth, G. K.edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
-
Nakamura, Y., Gojobori, T. & Ikemura, T. Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 28, 292 (2000).
-
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neubock, R. & Hofacker, I. L. The Vienna RNA websuite. Nucleic Acids Res. 36, W70–W74 (2008).
-
Tang, W. et al. Data and code related to ‘Prime editor-based high-throughput screening reveals functional synonymous mutations in human cells’. Zenodo https://doi.org/10.5281/zenodo.14639522 (2025).
Acknowledgements
We thank the National Center for Protein Sciences at Peking University for their assistance with fluorescence-activated cell sorting and analysis, especially H. Lv and H. Yang for their technical support. We thank the High-Performance Computing Platform of Peking University for enabling our data analysis. We thank the National Center for Protein Sciences PKU-EdiGene High-Throughput Screening Core at Peking University. We thank Tsingke Biotech for providing primer synthesis and sequencing services. We thank Cloud-Seq Biotech for providing the Ribo-seq service and Emei Tongde Technology Development for their technical support in single-cell RNA library preparation. This research was funded by the National Natural Science Foundation of China (31930016, to W.W.), the Peking-Tsinghua Center for Life Sciences (to W.W.), Changping Laboratory (to W.W.) and the Taishan Scholarship (tsqn202312362, to Yongshuo Liu).
Ethics declarations
Competing interests
W.W. is a scientific advisor and founder of EdiGene and Therorna. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
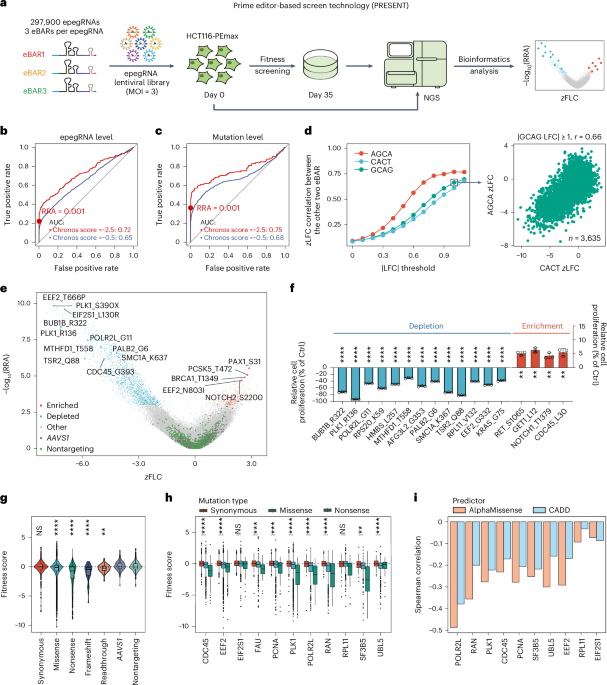
Extended Data Fig. 1 Design of the epegRNA library for screening functional synonymous mutations and development of the experimental system using PEmax.
a, Diagram of the epegRNAeBAR library structure. RTT: reverse transcription template, PBS: primer binding site. b, Principles and workflow for designing epegRNAs within the library. Searches for NGG PAMs are conducted on the representative transcript of each gene to determine the spacer sequence, PBS sequence, and RTT, followed by a first filtering step. Comprehensive searches along the coding region are then performed to achieve saturation mutagenesis of synonymous mutations, with linker sequences subsequently designed and a secondary filter applied, resulting in the final epegRNA sequences. c, Distribution of expression levels and essentiality of genes within the library in HCT116 cells. Red dots represent the 11 genes targeted for complete saturation mutagenesis, blue dots represent the 56 genes targeted for synonymous saturation mutagenesis, and gray dots represent all other genes. d, Histogram of the number of epegRNA designed for mutations in the library. e, Statistics of the types of amino acid substitutions for the 11 genes targeted for saturation mutagenesis. The y-axis represents the original amino acid, the x-axis represents the amino acid post-PE editing, and the values represent the number of epegRNAs designed for each substitution. The histogram on the right illustrates the coverage of the corresponding amino acids, indicating the proportion of amino acids that can be targeted among all amino acids in the 11 genes. f, Clustered heatmap displaying Pearson correlation coefficients of the whole transcriptome between HCT116-PEmax cells and wild-type (WT) HCT116 cells, as well as after the addition of nontargeting (NT) epegRNA and AAVS1-targeting epegRNA. g, Graph showing the changes in editing efficiency over a 28-day period of continuous culture in HCT116-PEmax cells with the addition of two test epegRNAs targeting different sites. The data is presented as the mean ± s.d. (n = 3 biological replicates).
Extended Data Fig. 2 Decoding and ROC analysis of the epegRNAeBAR library.
a, Diagram illustrating the genomic PCR process performed on the cell library post-screening. b, Algorithm description of ZFC-eBAR (see Methods for details). c, The x-axis represents the number of genes corresponding to the positive controls (epegRNAs that introduces nonsense and frameshift mutations on essential genes), while the y-axis represents the AUC calculated using these positive controls as well as negative controls (AAVS1-targeting and nontargeting epegRNA). The score used to calculate AUC is the screen score. d, Similar to c, but nonsense/frameshift mutations at the same site are regarded as the same mutation. The score used to calculate AUC is the Top score. e, ROC analysis of epegRNAs in highly essential genes (Chronos score < −2.5) based on different scores. The red point represents the threshold we selected. The red line represents the ROC curve for the top1 score of epegRNA, the dark blue line represents the ROC curve for the top2 mean score of epegRNA, and the green line represents the ROC curve for the all mean score of epegRNA. f, Spearman correlation of zLFC for the top 2 ranked epegRNAs in highly essential genes (Chronos score < −2.5).
Extended Data Fig. 3 Evaluation of screening reproducibility and detection sensitivity.
a, Correlation of zLFC values between two eBARs replicates for epegRNA with the third eBAR’s |LFC| ≥ 0. b, Correlation of zLFC values between two eBARs for epegRNAs with |AGCA LFC| ≥ 1 and |CACT LFC| ≥ 1. c, Pearson correlation analysis of LFC values between two eBARs across varying thresholds of absolute LFC values from the third eBAR. The x-axis indicates the absolute LFC threshold applied to each eBAR, while the y-axis represents the Pearson correlation coefficient between the remaining two eBARs. d, Correlation of LFC values between two eBARs for epegRNAs with the third eBAR’s |LFC| ≥ 1. e, Correlation of LFC values between two eBARs for epegRNAs with the third eBAR’s |LFC| ≥ 0. For panels a, b, d, and e, Pearson correlation coefficient (r) is labeled. N indicates the number of epegRNAs shown in these figures (for panels a and e, excluding those with zero sequencing read counts and black lines indicate density-based contours). f–g, Correlation analysis of enriched mutations in the HCT116 screen across three eBARs, based on LFC values (f) or zLFC values (g). Diagonal panels show histograms of value distributions, lower left panels show scatter plots of pairwise correlations, and upper right panels present Pearson correlation coefficient (r). The dataset includes 1,717 epegRNAs with nonsynonymous mutations and 417 epegRNAs with synonymous mutations. h, Boxplot of the relationship between the degree of reduction in counts of simulated deleterious variants in the experimental group (x-axis) and the calculated |screen score| (y-axis). n = 492 per group (number of epegRNAs). The gray dashed lines correspond to thresholds of 3 and 2.2. Boxplots are depicted as follows: the center line represents the median, the box limits denote the upper and lower quartiles, and whiskers extend to 1.5 times the interquartile range. i, Corresponding to h, the x-axis represents the average counts of these simulated deleterious variants in day 0, and the y-axis indicates the percentage reduction in day 35. Green labels highlight points where the |screen score| after reduction exceeds the selection threshold (3).
Extended Data Fig. 4 Performance of synonymous mutations in human homologous genes of the yeast.
a, Comparison of fitness score distributions for synonymous mutations across three groups: 67 saturation-mutated genes (synonymous mutations only), 11 saturation-mutated genes (including all mutation types), and human homologous genes of the yeast (excluding RPL39 due to limited data). P values were calculated using a two-sided Wilcoxon test. b, Distribution of essentiality for human homologous genes of yeast in HCT116 cells, data of RPL29 are missing in DepMap. The corresponding positions are labeled in purple. c, Shown in order as in b, distribution of fitness scores for synonymous, nonsense and frameshift mutations for each gene. From left to right, the mutation counts for each group are 46/5 (RPS7), 269/6 (TSR2), 459/6 (BUD23), 167/6 (ATP6V1F), 791/6 (PAF1), 233/6 (GET1), 131/6 (VMA21), 162/6 (LSM1), 193/6 (UBE2B), 323/6 (INO80C), 487/6 (PRPS2) and 80/6 (RPL29), with n = 494 for AAVS1 and n = 981 for Nontargeting. Boxplots are depicted as follows: the center line represents the median, the box limits denote the upper and lower quartiles, and whiskers extend to 1.5 times the interquartile range.
Extended Data Fig. 5 Comprehensive analysis of different mutation types and their effects in saturation mutagenesis.
a, Correlation of BLOSUM62 scores with the screening results for 11 genes. Mean scores were compared to nontargeting controls, with mutations showing adjusted P < 0.05 (two-tailed Student’s t test with Benjamini–Hochberg correction) marked in black. Only mutations with counts ≥10 were included. b, Upper panel displays a heatmap of screen scores for all mutations in RAN. Light gray indicates amino acids not included in the library. Synonymous mutations are marked with dots, and dashed lines denote domain boundaries. The lower panel illustrates domain annotations for RAN. c, Top scores for missense and synonymous mutations are compared across all regions of RAN, including G3 Switch II and Q69, n indicates the number of mutations. d, Comparison of top screen scores from our study with scores from ref. 22. Spearman correlation coefficient = 0.3076786. e, Detailed Mutation Analysis in POLR2L. Middle panel: Heatmap of screening scores for all mutations in POLR2L. Light gray indicates amino acids not designed in the screening library. Dots indicate synonymous mutations. Dashed lines represent four Zn2+ binding sites. Lower panel: Average top scores across each position, with three designed Zn2+ binding sites highlighted in red. Left panel: Average top scores by amino acid, with the two most depleted ones shown in orange. f, Cell fitness effects of different amino acid (aa) substitutions (statistical methods as a). g, The distributions of fitness scores for mutation types with a significant impact on cell fitness. From left to right, the mutation counts are 1,475, 182, 258, 113, 233, 230, and 850 respectively. h–i, Distribution of enriched mutations for various amino acid substitutions (h) and among different types of base pair substitutions (i) in both saturation synonymous mutations and clinical synonymous mutations. Top score represents the maximum |screen score| value for different epegRNAs corresponding to each mutation in the screen. For c and g, boxplots are depicted as follows: the center line represents the median, the box limits denote the upper and lower quartiles, and whiskers extend to 1.5 times the interquartile range.
Extended Data Fig. 6 Distribution of different key features for synonymous mutations.
a, Distribution of SpliceAI scores for synonymous mutations. b, Distribution of RNA folding energy changes for synonymous mutations. c, Distribution of codon usage frequency changes for synonymous mutations. d, Distribution of conservation scores for synonymous mutations. e, Distribution of gene essentiality (Chronos score) for synonymous mutations in HCT116 cells. f, Distribution of expression levels for synonymous mutations in HCT116 cells.
Extended Data Fig. 7 Validation of synonymous mutations causing aberrant RNA splicing.
a, Schematic depiction of the splicing alterations caused by the RPL11_V132 (GTG > GTC) mutation. The transcript sequence information was obtained by sequencing the cDNA from the experimental and control groups. b, Validation of the effect of the RPL11_V132 (GTG > GTC) mutation on cell proliferation in HCT116 cells. c, Relative mRNA expression levels of RPL11 in the experimental and control groups. The mRNA level of each sample was quantified by real-time qPCR and normalized by GAPDH, and the indicated relative mRNA level was normalized to that of AAVS1-targeting control cells. d, Analysis of editing outcomes for epegRNA targeting RPL11_V132 and controls via genome sequence amplification and NGS. e, Schematic depiction of the splicing alterations caused by the KRAS_G75 (GGG > GGT) mutation. The transcript sequence information was obtained by sequencing the cDNA from the experimental and control groups. f, Validation of the effect of the KRAS_G75 (GGG > GGT) mutation on cell proliferation in HCT116 cells. g, Relative mRNA expression levels of KRAS in the experimental and control groups. The mRNA level of each sample was quantified by real-time qPCR and normalized by GAPDH, and the indicated relative mRNA level was normalized to that of AAVS1-targeting control cells. h, Analysis of editing outcomes for epegRNA targeting KRAS_G75 and controls via genome sequence amplification and NGS. All data are presented as mean ± s.d. (n = 3 biological replicates for cell proliferation assay, n = 3 technical replicates for real-time qPCR). P values were calculated using two-tailed Student’s t test, *P < 0.05, ****P < 0.0001; n.s., not significant.
Extended Data Fig. 8 Editing and the gene expression analysis at the PLK1_S2 site.
a, Analysis of editing outcomes for epegRNA targeting PLK1_S2 and controls via genome sequence amplification and NGS. b, Relative mRNA expression levels of PLK1 in the experimental and control groups. The mRNA level of each sample was quantified by real-time qPCR and normalized by GAPDH, and the indicated relative mRNA level was normalized to that of AAVS1-targeting control cells. Data are presented as mean ± s.d. (n = 3 technical replicates). P values were calculated using two-tailed Student’s t test, n.s., not significant.
Extended Data Fig. 9 Capture and data quality control for DIRECTED-seq.
a, Overview of the process for capturing epegRNA and single-cell transcriptomes. The cDNA of epegRNA was obtained by reverse transcription using primers targeting the constant secondary structure of evopreQ1, and the cDNA of the entire transcriptome within the cells was obtained by reverse transcription using oligo-dT primers. The figure was adapted from ref. 63. b, Sequencing depth impact epegRNA detection probability and observed gene expression levels. The blue dashed line represents the linear regression line correlating total epegRNAs per cell (y) with total UMIs per cell (x). c–d, Quantile–quantile plots comparing 15 nontargeting epegRNAs (negative control) with 395 epegRNAs across 213 genes (discovery). Genes for the nontargeting tests were randomly selected from the entire gene set. The left-hand panel shows the QQ plot of raw P values, and the right-hand panel shows the QQ plot of –log10 transformed P values. In both panels, the black solid line denotes the theoretical y = x reference under the null hypothesis and the surrounding gray band represents the pointwise 95% confidence interval of that regression.
Extended Data Fig. 10 Comparison of the performance of different prediction models and the screening results in the A549 and KYSE-30 cell lines.
a–b, ROC analysis of DS Finder, SilVA, and CADD on 45 confirmed pathogenic mutations (positive controls) and 1,439 synonymous mutations without phenotypes from our screening (negative controls). The x-axis represents the false positive rate, and the y-axis represents the true positive rate. The red, dark blue, and green curves represent DS Finder, SilVA, and CADD, respectively. The test set in a contains 20 positive controls, excluding the mutations used in the SilVA training set and the test set in b consists of the 25 mutations used in the SilVA training set. c–f, Screening results analysis of three cell lines. Volcano plot illustrating the results of screening for functional synonymous and nonsynonymous mutations affecting cell fitness in A549 (c) and KYSE-30 (e). Blue and red dots denote depleted and enriched epegRNAs, respectively. Analyzing the correlation between three eBARs using zLFC in A549 (d) and KYSE-30 (f), following the same method as in Fig. 1d. The data were filtered according to the log2 fold change of a specific eBAR and the Pearson correlation of the zLFC of another pair of eBARs was investigated. The y-axis represents the Pearson correlation of the zLFC, while the x-axis indicates the specific threshold that the absolute value of the LFC must surpass. Red, green, and blue denote three individual eBARs: AGCA, CACT, and GCAG, respectively. g, Performance of DS Finder in three cell lines compared with CADD and SilVA. Each point on the graph represents a different dataset, totaling 10.
Supplementary information
Supplementary Information
Supplementary Figs. 1–5 and captions for Tables 1–10.
Reporting Summary
Supplementary Tables 1–10
Supplementary Table 1. The genes involved in the epegRNA library used for this screening. This table provides detailed information on these 3,644 genes and our selection criteria. Supplementary Table 2. Information about the epegRNA library. Sheet 1 provides an overall summary of the library. Sheet 2 contains detailed information, including the target gene, mutation details and sequence information for each epegRNA. Supplementary Table 3. Screening results in HCT116 cells. Supplementary Table 4. Single-cell screening results in HCT116 cells. Supplementary Table 5. Features of synonymous mutations. This table details 75 features used in the synonymous mutation analysis, with comprehensive presentation of the HCT116 screening results. Supplementary Table 6. Clinical prediction results of the DS Finder model and mutation information used for model analysis. Sheet 1 contains prediction scores of colonic disease-associated synonymous mutations by DS Finder. Sheet 2 includes 45 known clinically pathogenic synonymous mutations used for evaluating model prediction performance. Sheet 3 provides same-site controls for these 45 mutations. Sheet 4 contains negative control mutations obtained from screening. Supplementary Table 7. Screening results in A549 cells. Supplementary Table 8. Screening results in KYSE-30 cells. Supplementary Table 9. Sequence information of all epegRNAs used in validation experiments. Supplementary Table 10. All primers used in this study. Each sheet sequentially includes library PCR primers, genomic PCR primers for each validation site, cDNA PCR primers and reverse transcription–qPCR primers.
Source data
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Niu, X., Tang, W., Liu, Y. et al. Prime editor-based high-throughput screening reveals functional synonymous mutations in human cells.
Nat Biotechnol (2025). https://doi.org/10.1038/s41587-025-02710-z
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41587-025-02710-z










Related Posts



Stay Informed With the Latest & Most Important News
Previous Post
Next Post


Advertisement