Now Reading: Beyond cell atlases: spatial biology reveals mechanisms behind disease
-
01
Beyond cell atlases: spatial biology reveals mechanisms behind disease
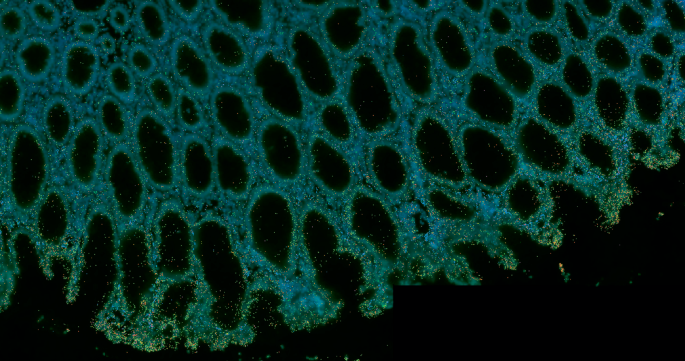
As the next generation of spatial transcriptomics tools hits the market, researchers are uncovering previously unknown interactions that could transform clinical research.

Vizgen
To deeply investigate cell–cell interactions, particularly at the frontiers of disease, researchers increasingly are turning to spatial biology. Single-cell RNA sequencing reveals much about the activities of individual cell types, but key interactions are lost by dissociating cells from their native configuration in tissues. Conversely, methods that retain spatial information have so far been limited in how much of the transcriptome they can capture at single-cell resolution. Lower-plex methods can provide cell type and location information, allowing researchers to make inferences about cellular interactions, but may miss details about the exact molecular mechanisms involved.
Now, a new era is dawning. Improved methods for spatial transcriptomics are becoming available that are capable of whole-transcriptome profiling at single-cell resolution. “I think a lot of people have come into spatial thinking of the next gold rush,” says Rob Tarbox, vice president of product and marketing at Complete Genomics. “‘Single-cell 2.0’ is kind of how I see it.”
In the five years since Nature Methods named spatially resolved transcriptomics its Method of the Year1, the field has matured considerably, and several companies have recently announced new or improved spatial transcriptomics products that raise the bar for performance. Many of these expand the applicability of spatial transcriptomics to clinical research, such as improving performance in formalin-fixed, paraffin-embedded (FFPE) tissue samples, increasing resolution, and lowering costs. Yet, in many ways, spatial transcriptomics remains in its infancy, with bioinformaticians and data scientists generating new analysis tools as fast as they can to keep up with the flood of data. “If you look at GitHub, literally every week there’s three new spatial tools because the field is evolving so fast,” says Jasmine Plummer, the director of the Center for Spatial Omics at St. Jude Children’s Research Hospital. Among the challenges of the field overall are the lack of standardization in analysis parameters and the fact that different commercial platforms generate different file formats.
Other obstacles to bringing spatial transcriptomics to the clinic include high turnaround times, high costs per sample and a lack of regulatory frameworks for validating spatial transcriptomics as clinical diagnostic tools. “Proteomics is closer to the clinic purely because pathologists can interpret proteins a lot easier than they can RNA,” says Arutha Kulasinghe, who leads the Clinical-oMx Lab at the Frazer Institute, University of Queensland, in Brisbane, Australia. “It just makes sense in terms of the interpretability of the signal, the robustness of antibodies and things like that. But it’s not to say that we don’t need the spatial transcriptome, too.”
Is spatial transcriptomics worth it?
Running a spatial transcriptomics experiment is expensive and time-consuming. The run itself can take up to a week, and analyzing and interpreting the vast amounts of data generated are not trivial processes. “We’re all excited by new technologies, but I, as a lab head, know that if we generate a dataset today, I’m probably at least 12 months away from understanding what we’re looking at,” says Kulasinghe. Given that, it is unwise to foray into spatial transcriptomics unless you have a clearly defined biological question that cannot be satisfactorily addressed by simpler or less expensive methods. When are spatial transcriptomic data really necessary?
“Where transcriptomics really shines is understanding mechanism,” Kulasinghe says. “Mechanisms driving resistance, mechanisms driving sensitivity, we can’t really tease that out from the protein data.” Kulasinghe studies cancer, and one critical area of research right now is understanding why many cancers do not respond to immunotherapy. Often, patients whose disease is resistant to checkpoint inhibitors have less immune infiltration of their tumor, but it is not apparent why that happens. “At the protein level, all we could say was, ‘It’s less infiltrated’,” Kulasinghe says. The RNA data revealed that immunotherapy-resistant tumors had shifted their energy metabolism in ways that prevent immune cells from infiltrating the tumor, and understanding these metabolic changes could reveal new drug targets, Kulasinghe says. “What we need to understand at a mechanistic level is what pathways in the tumor or the immune cells are being turned on or off, and we can only understand that at the RNA level.”
Why certain therapies work in some patients and not others remains a major question, not just for immunotherapy but many other diseases as well. Parambir Dulai is director of precision medicine for the Division of Gastroenterology and Hepatology at Northwestern University in Chicago, and a gastroenterologist specializing in inflammatory bowel disease. Drugs for this condition work about a third of the time, Dulai says, and even in cases where the disease appears clinically similar, patients sometimes have different responses to drugs. “It’s been extremely difficult to be able to define the specific mechanism that’s driving the inflammation for an individual patient,” he says. Using spatial transcriptomics, “what we’re doing is we’re essentially taking decades of pathology research and elevating it now to link all of that to the underlying mechanisms that might be contributing.”
Dulai and his colleagues successfully identified a particular myeloid cell–fibroblast interaction that was contributing to non-response to a drug. That interaction could lead to a targetable pathway or a diagnostic test to identify patients whose disease is unlikely to respond to the drug. To find out, the researchers are analyzing tissue from patients who have been treated with different drugs, comparing their pre- and post-treatment samples, and investigating how gene expression correlates with response or non-response to the drug. “The ability to apply spatial transcriptomics technology to banked FFPE tissue blocks offers us an ability to find patients,” Dulai says. “We can go back in time and collect these archived samples and create really nice cohorts of very well-matched patients that would have otherwise taken a decade to recruit.” The findings could drive drug development, Dulai says, or suggest combination strategies of existing drugs that act on different pathways, sidestepping resistance and creating longer-lasting treatments.
Exploring the borderlands
The boundaries between disease and healthy tissue may reveal hidden signs of disease that are not apparent from visual inspection or detection of protein biomarkers. For example, in pulmonary fibrosis (PF), “We might have people who are very stable for a long period of time and then people who are going to progress really rapidly, and right now, we don’t really know what’s driving people to walk down one of these different trajectories,” says Nicholas Banovich, director of the division of bioinnovation and genome sciences at the Translational Genomics Research Institute (TGen) in Phoenix, Arizona. “We are trying to understand the molecular changes that differentiate outcomes, and having this really clear spatial information can help us dissect that much better than even with what we could do with single-cell.” With dissociative single-cell RNA sequencing, Banovich says, cells that are fragile or deeply embedded in extracellular matrix may not be represented in the data. “Fibroblasts, which are obviously a key component of PF, tend to not come out very well in our single-cell datasets,” Banovich says. “If I have a heterogeneous piece of tissue, where some pieces of it are less diseased and some pieces of it are more fibrotic or more remodeled, when I do a single-cell dissociation I’m not actually getting equal representation of cells across all of my disease features.” To better capture the specific expression patterns of cells on the boundary between disease and normal tissue, he says, requires spatial transcriptomics.
Indeed, when they overlaid RNA data onto histology data in lung samples from patients with pulmonary fibrosis, Banovich and colleagues observed that some lung cells that appeared normal showed significant molecular dysregulation2. Understanding the molecular changes that precede cellular damage could provide insight into why some patients’ disease progresses faster — and how to slow it.
Similarly, the cell–cell interactions around the edges of a tumor can spell the difference between a slow-growing tumor and an aggressive, deadly cancer. Tumor microenvironment dynamics influence the progression of a premalignant lesion to a stage 1 cancer, stage 1 to stage 2, and so on3. “There is an organization in the chaos of those cells. It’s not a random process,” says Kulasinghe. A detailed map of the interactions that accompany the transition from early-stage to advanced-stage disease could yield information about how to interfere with that organization. “If we prevent those cells from coming together in the first place, can we slow the progression of that cancer?”
When RNA is not enough
In some cases, the transcriptome shows only part of the picture. “Because we are interested in neurodegenerative diseases that accumulate proteins, particularly tauopathies and Alzheimer’s disease, there’s a need for proteomics,” says Miranda Orr, a neuroscientist at the Washington University in St. Louis School of Medicine who studies cellular senescence and neurodegenerative disease. Tau protein, which forms neurofibrillary tangles associated with neurodegenerative disease, exists in several different forms. Orr’s team uses spatial transcriptomics and proteomics methods to measure gene expression of tau and protein expression of multiple tau protein forms together, individual levels of three different splice variant groups and eight differently phosphorylated forms of the protein. The researchers compared neighboring neurons where one had tau accumulation and the other did not. “The RNA level in neurons that have pathogenic tau is lower than the RNA level of neurons without pathogenic tau,” Orr says. “If you’re only looking at transcriptomics, you would not have any way of knowing that those cells that have downregulated tau at the gene level actually have loads of protein deposited inside of them.”
Spatial proteomics kits have improved considerably from just a few years ago, when imaging 84 proteins was standard. Today, Orr says, she can image 1,100 proteins in a single sample. “You start to gain greater insights into what’s happening with that molecule in cells that are undergoing some pathogenic process versus those aren’t,” she says. “If you were only interpreting based on the RNA, you would have a very, very different interpretation than if you’re looking at protein, which would be very different than if you’re looking at post-translational modifications.”
New products hitting the market
As the technology matures, companies are rolling out new capabilities that will improve researchers’ ability to ask clinically important questions. Analyzing banked tissue samples, for instance, offers substantial advantages in terms of study design, but the RNA may have degraded. “It’s common that the academic research community or pharma community, people who want to do these types of measurements, often don’t always have samples where the RNA is perfectly intact,” says George Emanuel, scientific cofounder and vice president & portfolio owner, instruments at Vizgen, the company that commercialized the technique MERFISH.
Vizgen has just rolled out MERFISH 2.0, which has increased sensitivity for fragmented RNA molecules. MERFISH imaging uses multiple probes tiled along the length of a transcript, and MERFISH 2.0 enhances sensitivity by improving how the transcript fragments are anchored in the gel. That way, when the probes bind to the fragments, they remain colocalized and all the fragments contribute to the intensity of the spot, explains Emanuel. Other improvements include higher-intensity probes and enhanced hybridization conditions to boost the signal.
Last year, Vizgen had customers send in samples they found difficult to image using the original MERFISH protocol and ran them with MERFISH 2.0. “We saw some great improvements on a whole variety of different sample types, for some improvements up to tenfold in sensitivity with MERFISH 2.0 relative to the MERFISH 1.0 results they were getting,” Emanuel says. “It’s an enhanced chemistry that improves sensitivity across the board, but in particular on samples where the RNA has degraded.”
Spatial transcriptomics techniques generally fall into two categories: imaging-based, like MERFISH, or sequencing-based. The sequencing-based approach can capture the whole transcriptome in an untargeted manner, but so far has provided lower-resolution data than imaging-based approaches. Soon, however, it may not be necessary to choose between coverage and resolution, as companies develop advanced products to address these gaps.
In February Illumina launched its new spatial transcriptomics kit, which offers sequencing-based, untargeted whole-transcriptome analysis with cellular resolution. “The two things we thought were needed were unbiased whole transcriptome, so the ability to look for whatever you want, combined with cellular resolution,” said Darren Segale, senior director of scientific research at Illumina. “That is kind of missing in the market today — you have to choose one or the other.”
The system will run on Illumina’s NovaSeq or NextSeq sequencers and include a library kit, as well as analysis software via Illumina Connected Multiomics. It can accommodate a larger sample area than any platform currently available, at 50 mm × 15 mm. Illumina promises fourfold higher resolution and fourfold lower cost per unit area compared to existing methods. The product will be available commercially in 2026, although several early access customers are already getting data. (These users did not work directly with the kit, but sent their samples to Illumina for processing, then received the data back.)
“It is a very large capture area, so I think that’s their biggest advantage,” says Banovich. “It’s a massive step up in size. Throughput is really a significant factor for the size and scope and scale of the studies we can do, so having that much larger capture area, with pretty good sensitivity for a whole transcriptome assay — those two things together are probably the most exciting piece of that platform.”
Imaging-based methods generally capture single-cell or subcellular resolution but rely on preselected panels of probes for detection. Now Bruker is introducing a whole-transcriptome (WTX) panel for their CosMx Spatial Molecular Imager, slated for commercial release in summer 2025. The panel includes 37,872 imaging barcodes, and the company claims subcellular imaging of the entire human protein-coding transcriptome. At the Advances in Genome Biology and Technology (AGBT) general meeting in February, Bruker presented data showing that WTX performed as well as droplet-based single-cell analysis technology in terms of detection efficiency, with superior detection of irregularly shaped epithelial cells and detection of rare cells, such as endocrine epsilon cells4.
CosMx also allows simultaneous RNA and protein imaging on the same slide, which can reveal more detail about, for instance, tumor behavior. In his talk at AGBT, Joseph Beechem, CSO and senior vice president of Bruker Spatial Biology, presented a non-small cell lung cancer slide in which the transcriptomics showed big empty spaces between the tumor areas and the immune infiltrate. Overlaying the proteomics data revealed that those gaps were actually thick layers of extracellular fibronectin and smooth muscle actin, generating a barrier between the tumor domain and the immune cells. “I love RNA, but it doesn’t give you the whole story,” Beechem said. “I’m looking at that transcriptome and I want to put it in the proper context.”
Complete Genomics, the distributor of STOmics Stereo-seq technology in the United States, recently announced two new specialized kits, one for processing FFPE tissue and one large-chip kit. “Our FFPE product is a one by one centimeter chip that allows you to use randomers,” says Tarbox. “It’s not targeted.” The large-format chip requires fresh frozen tissue but increases the size of the sample area to two by three centimeters. This month, Complete Genomics awarded four grants through a program called Spatial Xcellerator to get the new products into the hands of US-based researchers whose projects will particularly benefit from these advanced capabilities. The program offers free reagents and sequencing, as well as training and support covering the entire process, from sample preparation to data analysis. The projects that received awards address topics including pediatric brain tumors, spinal cord injury and Alzheimer’s disease.
Interpreting the data remains a challenge
Complete Genomics also recently announced a partnership with BioTuring, a developer of bioinformatics algorithms and software. BioTuring’s SpatialX is a deep-learning tool for analyzing spatial data analysis, and within SpatialX there are preoptimized pipelines for analyzing data from STOmics products. “BioTuring has been in this space for quite a while,” says Tarbox. “I think from a software flow perspective, it’s a natural progression to take the output from our software; the data files are standard, and you’re leveraging a tool that has already been built and used for other technologies.”
Indeed, while most commercial spatial transcriptomics platforms include some analysis software, users generally look to specialized software for analyzing their data, either via GitHub or through purchase from a software provider. Companies such as BioTuring, Rosalind, Predx, Elucidate and others are filling that gap, often leveraging artificial intelligence (AI) capabilities to speed up, automate and increase the accuracy of analysis tools, such as with improved cell segmentation. Labeling the identities of all the cell types in a region and the state they are in, however, is just the first step in mining transcriptomics data, explains Ken Bloom, head of pathology at Nucleai, an AI-focused spatial biology company. “The secret sauce is really to understand all the relationships between those cells and how they interact with each other,” he says. “We try to provide insight through AI as to how they interact and the meaning of that interaction.” This is particularly useful when coupled with information about how a condition has responded to therapy, for example, to create a more detailed picture of what cellular and molecular processes contribute to resistance or response to particular drugs. Understanding how different cells are communicating and influencing each other relies on detailed identification of all cell types in the sample, something that some analysis algorithms struggle with.
“Imagine somebody takes a picture of you, but there’s somebody standing next to you, and they put their hand on your shoulder,” Bloom says. The other person isn’t visible in the photo, but someone looking at the photo would know it’s most likely the hand belongs to a second person who is out of frame, not that the hand is growing out of your shoulder. “That happens with cell types,” Bloom explains. “You might have a tumor cell, and there might be a macrophage that’s like a ‘hand on its shoulder,’ so when you stain with a macrophage stain, the tumor cell now looks like it’s expressing that macrophage marker.” Bloom says that Nucleai’s classification system has learned how to better distinguish these metaphorical ‘hands on shoulders’ and correctly interpret that two cells are interacting. “That could be a critical relationship,” Bloom says. “Other systems throw it away. They confuse it. It’s not just that they don’t understand that the hand was on your shoulder — they might not even recognize you as you anymore.”
Once the cell types and interactions are defined, the next step is to tease out how those interactions predict and influence clinical outcomes. “We’re still in the discovery phase of how you take that complex information, marry it with AI to understand the interactions, and then marry that with clinical outcome data, so we understand which patterns and which interactions are necessary for therapies to work,” Bloom says.
Advanced AI tools could bring down costs, speed up analysis and widen the practical applications for spatial biology. In a recent preprint, Faisal Mahmood and colleagues at Harvard Medical School reported a new AI framework called VORTEX that can predict gene expression in a 3D tissue sample5. VORTEX, which stands for volumetrically resolved transcriptomics expression, combines 3D tissue morphology with 2D spatial transcriptomics to resolve the transcriptome of the entire 3D sample.
Obtaining 3D spatial transcriptomics by serial sectioning can be prohibitively expensive. “Depending on which technology you use, each individual section could cost up to $5,000,” Mahmood says. “By the time you’re done with a three-dimensional block, it could be close to $100,000 or even more.” VORTEX brings down the cost because it requires transcriptomics from only one or two sections. The AI is pretrained on large datasets that include 2D spatial transcriptomics with the corresponding histology data, teaching it the features of the tissue type. “The reason this works is that there has been so much of that data already collected and in the public domain,” Mahmood says. “There have been hundreds of studies doing spatial transcriptomics on basically every organ.” The model is then fine-tuned on data from the tissue being tested, teaching it how to correlate the morphological and spatial transcriptomic data.
Having 3D gene expression data could improve clinical diagnostics, Mahmood says. “There are a number of studies in pathology showing that as you cut through the block, your diagnosis can change,” he says. For instance, when determining something like Gleason grade in prostate cancer, the evaluation may be different depending on what features the tested slice happens to capture. “It makes sense to us that using an entire 3D volume would allow us to make a better, more accurate diagnosis.”
Seeing what has been missed
As increasingly sophisticated transcriptomic and proteomic kits come on the market, enabling visualization of a dizzying array of proteins and RNAs, bioinformaticians and computational biologists are rising to the occasion, turning the mountains of data into actionable findings. While there are still challenges to overcome before spatial multi-omics become as routine as genome sequencing, biomedical researchers are embracing this powerful technology, and the way forward will include a combination of techniques. “In general, scientists recognize the importance of understanding multiple levels of biology, whether it’s DNA, RNA, protein, post-translation modifications or microRNAs,” says Orr. “While single-cell and single-nucleus sequencing provide powerful insights, they lack spatial context; we don’t see where those transcripts are within cells or tissues. We don’t know where those cells were in space. We’re just missing so much information.”










Related Posts


Stay Informed With the Latest & Most Important News
Previous Post
Next Post



Advertisement