Now Reading: Powering new therapeutics with precision mitochondrial editing
-
01
Powering new therapeutics with precision mitochondrial editing
More precise base editors make mitochondrial DNA editing efficient enough to model disease and correct pathogenic mutations in rodents, but they are slow to move into clinical trials.
Mitochondrial diseases are complex metabolic disorders caused by mutations in either the mitochondrial or the nuclear genome that affect the body’s ability to generate energy. The diagnosis and treatment are challenging — neurological and non-neurological symptoms are not consistent between patients and depend on which tissues and organs carry a particular mutation. Conventional therapies are largely organ-specific, targeting commonly affected tissues with high energy demands such as the brain and muscles, and focus on alleviating symptoms or modifying mitochondrial function using small molecules, metabolic reprogramming or mitochondrial replacement therapy1. None of these treatments are curative.
Therapies correcting permanent mitochondrial DNA mutations are needed, as mitochondrial diseases are among the most prevalent inherited disorders2,3. Recent studies are making strides in addressing this, using engineered, precise mitochondrial base editors that could permanently correct pathogenic mitochondrial mutations. However, as base editing and epigenome editing approaches for modifying the nuclear genome move into late-stage clinical trials, mitochondrial genome editors are progressing more slowly into the clinic.
While there is a substantial overlap of the limitations of nuclear and mitochondrial genome editing — including off-target mutations, systemic and targeted delivery, and immunogenicity of the delivered editing components, there are also differences and unique challenges.
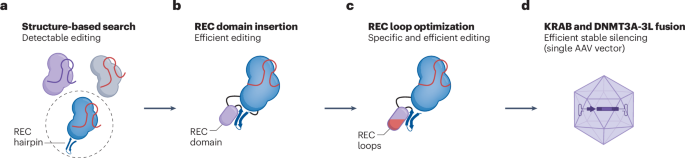
A major hurdle is the delivery of the editing machinery to the mitochondria. Mitochondria transport nucleic acids inefficiently, including the guide RNA that is required for CRISPR systems to operate4. Instead, CRISPR-free alternative nucleases that can be delivered as proteins are used, such as TALENs (transcription activator-like effector nucleases) and zinc fingers (ZNFs). Current mitochondrial DNA base editors generally comprise a CRISPR-alternative DNA-targeting domain (TALEN or ZNF) and a DNA editing domain for changing cytidine (C) to thymine (T) or adenine (A) to guanine (G). A short signal peptide added to the proteins guides them through the mitochondrial membrane. Cytidine base editors (CBEs) commonly use a TALEN (in fewer cases, a ZNF), the deaminase domain of bacterial toxin DddA (or APOBEC), and an uracil glycosylase inhibitor to avoid excising the nucleotide completely, instead of replacing it. Like CBEs, adenine base editors (ABEs) generally comprise a custom-designed TALEN and, in this case, a TadA deaminase variant that modifies the nucleotide base of interest5.
Like CRISPR editing, this process is susceptible to potential errors. The substantial off-target rate in mitochondrial and nuclear DNA is partly due to the design of the editors themselves6,7. TALENs and ZNFs are prone to nonspecific DNA interactions, which can cause unwanted deaminase activity at non-target sites, leading to bystander editing within an area around the actual target site. Several engineered editors exist that are more specific; however, depending on the target site, unwanted byproducts may be minimized but not eliminated8. CRISPR systems can be easily adapted by reconfiguring the matching single guide RNA (sgRNA), but ZFNs and TALENs require more extensive engineering for each new target site, making them less suited for clinical applications that require flexibility9,10.
Additionally, each cell contains hundreds to thousands of mitochondria carrying multiple genomes. The challenge is not simply to edit a single genome with two copies, as in nuclear editing, but to modify numerous cellular genomes simultaneously. Moreover, different populations of mitochondria can coexist within the cell, carrying mutated or wild-type mitochondrial DNA — a condition known as heteroplasmy. A specific threshold must be reached to correct pathogenic mutations in enough mitochondria for a phenotypic effect. Making it more difficult, the deaminases favor certain sequence contexts over others, limiting their general target range and further complicating their engineering. Although editing systems have been developed with expanded targeting windows for CBEs and ABEs8, their efficiency does not yet meet this threshold.
Progress is being made. Recent publications have demonstrated increased precision and efficiency of evolved base editors, allowing them to surpass the heteroplasmy thresholds necessary for generation of phenotypes and to model mitochondrial disease in rodents11,12,13.
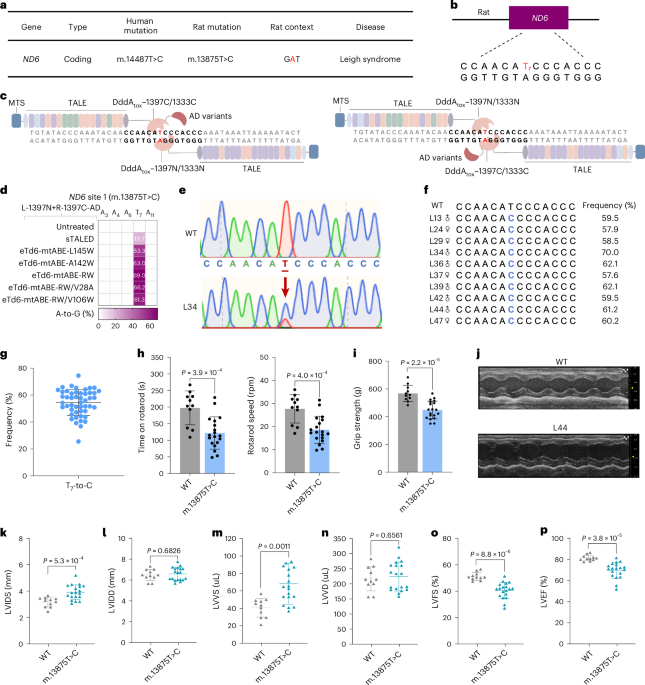
Animal models for mitochondrial disease are rare as a result of technical complexity and high costs, yet they are vital for mechanistic studies and testing of treatments14. Zhang and colleagues in Nature optimized mitochondrial base editors to target 70 mouse mitochondrial DNA mutations analogous to human pathogenic variants, demonstrating up to 82% efficiency in mice and 100% editing in the F1 generation, with minimal off-target edits11. Two major mitochondrial diseases — Leigh disease and Leber’s hereditary optic neuropathy — were modeled in the mice. Two more studies published this month in Nature Biotechnology12,13 generated rat models of Leigh disease and corrected disease-causing mutations using an evolved CBE, ameliorating the phenotype. These studies demonstrate a huge leap forward in the generation of animal models for mitochondrial disease and highlight the potential of engineered base editing systems to precisely edit nucleotides in mitochondrial DNA. Future work will hopefully take this from rodent to larger animal models, one more step toward clinical application.
Despite this progress, the full clinical potential of these base editors has not been unlocked. As in other editing technologies currently moving into the clinic, specificity and safety need to be tightly controlled. Additional modifying enzymes that could be used as an alternative to error-prone and bystander-heavy deaminases may broaden the targeting range while reducing specific engineering. Deaminase-free glycosylase-based editors that eliminate the off-target effects associated with deamination reactions and expand the range of base changes achievable have been explored for nuclear base editing15,16. These could be adapted for mitochondrial G-to-T base editing.
The three studies above used mRNA delivery via zygote injection, which is suitable for animal model generation. However, its clinical translation into therapy precludes postembryonic treatment. Somatic delivery to adult tissues remains a major hurdle. For some mitochondrial diseases affecting the eye, brain or muscle, local injections of adeno-associated virus vectors may be feasible. However, systemic delivery and access to hard-to-reach tissues, such as the central nervous system, are more challenging, as has been clear with nuclear editing systems. Ongoing efforts aim to reduce editor size for better packaging and delivery, as less accessible tissues are commonly affected by mitochondrial disease phenotypes17.
Technical barriers are progressively diminishing — a promising move toward possible clinical treatments — but it is still early. While biotechnologies from startups for nuclear genome editing are progressing through clinical development, the field of mitochondrial editing remains relatively quiet. Overcoming technical constraints is only the first hurdle. Investment, research and development costs, as well as regulatory guidelines make it difficult to continue moving forward — struggles CRISPR editing companies are currently facing. As the technologies themselves advance, it is anticipated that funders and regulators will keep up.










Related Posts



Stay Informed With the Latest & Most Important News
Previous Post
Next Post


Advertisement